Personalised medicine, unmet need or business strategy?
Abstract
Personalised medicine is a discipline that integrates pharmaceutical development with existing knowledge about the genetic and phenotypic factors that influence drug response. This integration enables tailoring therapies to individual patient’s characteristics to help improve their safety and efficacy. Personalised medicine implies a shift in the way drugs are delivered as it requires new forms of testing that assess the patient´s eligibility for a drug. This poses new challenges, both at a regulatory and reimbursement level.
Personalised medicine has reached higher commercial success and clinical uptake in drugs under development than old and off-patent drugs. This paper uses the case of TPMT testing to illustrate the reasons why personalised medicine for offpatent drugs is less used at a clinical level than personalised medicine for drugs under development.
1 Introduction
Personalised medicine (PM) is new approach to health care. It focuses on the study of the differences in drug response among group of individuals who share common genetic or phenotypic characteristics (Lindpaintner, 2003).
PM has improved the understanding of disease and drug response and is enabling better approaches to target discovery and drug development (Shah, 2004). Response to drugs has a genetic and/or phenotypic component and genetic and/or phenotypic variations among groups of individuals determine how they respond to drugs (Evans and Johnson, 2001). These differences are identified through biomarkers that (when transformed into companion diagnostics) have the potential of improving the safety and efficacy of both licensed drugs (blockbuster drugs) and drugs under development (minibuster drugs) (Lewis, 2003, Webster et al., 2004). For this, PM has been defined by two technological trajectories illustrated in figure 1). The first one focuses on safety, the second on efficacy (Hedgecoe and Martin, 2003).
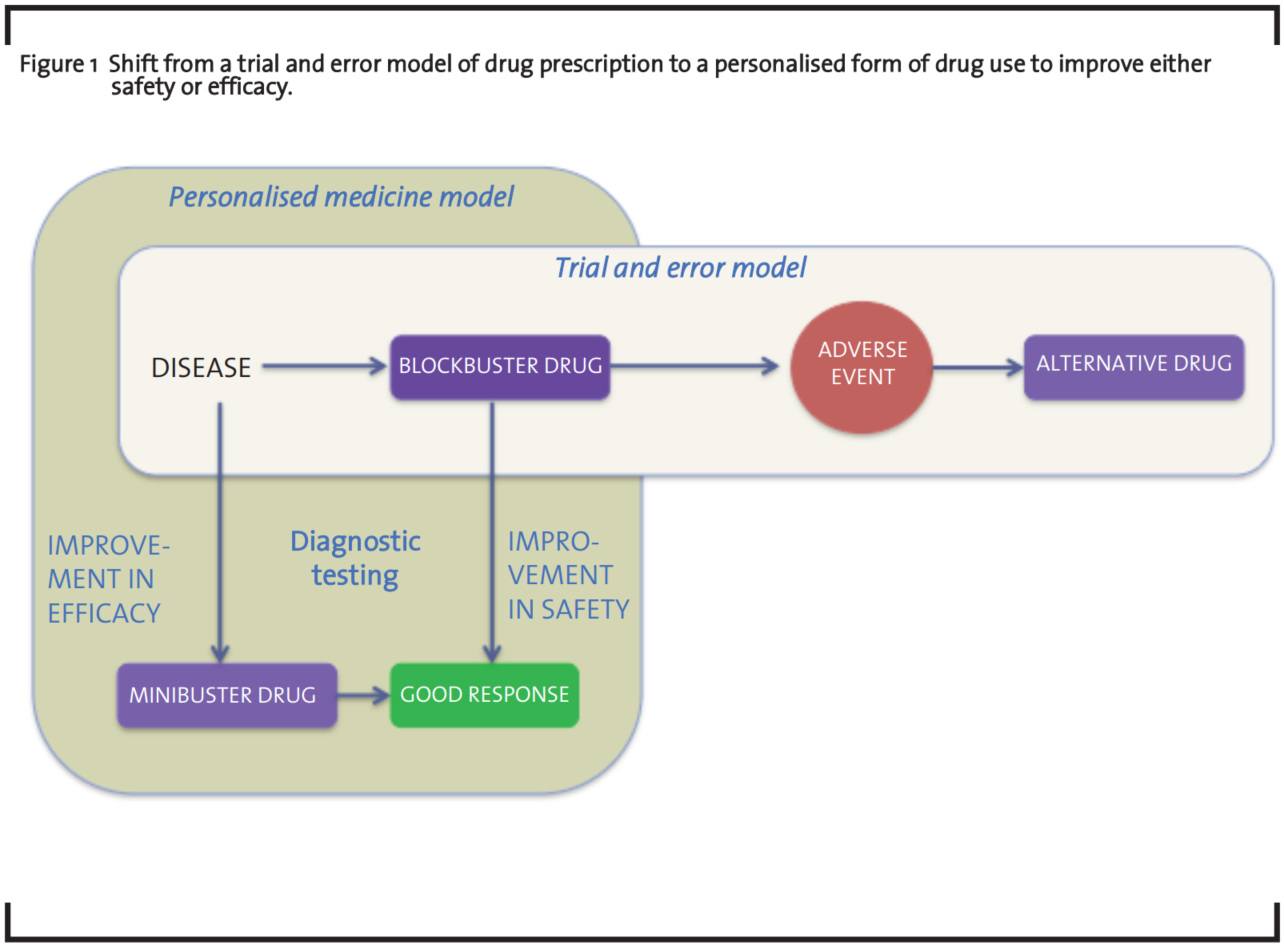
However, despite the promises around PM, few companion diagnostics have been launched to the market, most of these directed to drugs under development (i.e. Gleevec® in Philadelphia chromosome-positive chronic myelogenous leukemia, Herceptin® in HER2-positive breast cancer or Iressa™ and Tarceva™ in EGFR-positive lung cancer). Until now, the pharmaceutical industry has been more focused in the use of PM to drugs that are being developed rather than to drugs which are 10-50 years old (Human Genetics Commission, 2002).
Pharmaceutical companies urge to find strategies to face the decrease in the number of chemical entities to market. Despite the increase in research and development budgets over the last decade, the drug industry has been suffering a decrease in the number of marketing applications and approvals (Arnold and Hall, 2005). An increasing number of failures during regulatory approval, problems in the characterizing drug-dose effects and difficulties in measuring the risk-benefit ratio of new drugs have been at the origin of the problem (Di Masi et al., 2003). The increasing number of failures during regulatory approval, problems characterising drug-dose effects and difficulties measuring the risks benefit ratio of new drugs have accelerated the decrease of chemical entities to market (Di Masi et al., 2003). In addition, the constraints on clinical development due to a higher burden from regulators, the need to develop complex products with longer clinical development processes to ensure effectiveness in chronic patients and special sub-populations, and the need to carry these trials globally were some of the additional barriers in drug development (Milne, 2002).
Any new medicinal compound entering Phase I clinical trials have only an estimated 8% chance of reaching the market (FDA, 2004) and, even after approval, drugs may be withdrawn because of safety concerns. From 1990 to 2006, 38 drugs were withdrawn from major markets due to safety problems (Shah, 2006). Between 1998 and 2002 the average annual number of new drugs approved by the FDA was 68, by 2003, this number had dropped by two-thirds. In 2004 the number of approved drugs was 21 (Need et al., 2005).
2 Scope, research questions, and hypothesis
This paper analyses, retrospectively, the technological trajectory of TPMT testing and the process of clinical uptake by the UK National Health Service (NHS) until 2009. The purpose of the TPMT case study is analysing the main enablers and barriers during the introduction of TPMT testing in the NHS as well as the major hurdles associated to the process of reimbursement. In particular, the paper addresses the following questions:
- What were the main drivers and barriers that facilitated and/or hindered the use of TPMT testing in the NHS?
- How have these drivers and barriers shaped the process of technology diffusion? These questions are answered assuming the following hypothesis:
- The use of companion diagnostics or pharmacodiagnostics for improving the safety of offpatent drugs in the UK is less formalised than the use of companion diagnostics to improve the efficacy of drugs under development, because the reimbursement system for pharmacodiagnostics in the UK, is not designed to evaluate the unmet need of diagnostic or pharmacodiagnostics tests but their business potential.
3 Conceptual Framework
The conceptual framework used in this case study lies in three bodies of literature: diffusion of innovations theory and user-producer interactions to explain the process of technological diffusion and, socio-technical systems to explain the context of the diffusion.
3.1 Diffusion of innovations theory
The adoption and implementation of innovation has been traditionally explained through concepts addressed in the diffusion of innovation literature, which classical approach was developed by Rogers (Rogers, 1962). This model understands diffusion as the spread of ideas, mainly by imitation, with a special emphasis on the influence of social networks and how opinion leaders and individuals take adoption decisions. This concept stands on five principles: (1) the degree to which the innovation is perceived as being better than the previous one (relative advantage); for instance whether it represents an advantage in effectiveness and costeffectiveness; (2) the extent to which it is perceived as being consistent with the existing values, professional norms and ways of working (compatibility); (3) the complexity of the innovation, the barriers that need to be overcome and its difficulty of being used; (4) the possibility of experimenting with the innovation (trialability) and (5) the degree to which its results are visible to the intended adopters (observability). Innovation would be more easily adopted if the potential adopters could adapt and modify (or “reinvent”) the innovation to suit their own needs and innovations would be more rapidly “diffused” the more they complied with these principles.
This model was adapted by medical sociologists to explain the introduction of tetracycline in clinical practice (Coleman et al., 1966). However, Rogers’ five step unidirectional rule is not applicable to medical innovation for various reasons.
The diffusion of medical technologies involves a variety of actors and institutions, who need to align their differing interests through a process of collective social learning that will lead to the acceptance of certain technological trajectories and the rejections of others (Dosi, 1982).
Technological change in medicine relies on a series of feedback mechanisms among users and producers, who engage in a series of interactions that contribute to the re-shaping of the innovation (Gelijns and Rosenberg, 1994). In addition, medical innovation is highly regulated, both at the point of development and delivery and these regulations are also involved in these feedback mechanisms.
According to Rogers, consumers are the final users of the innovation. However, in the case of medical innovation, clinicians are often the gatekeepers of treatments and, in effect, the final users.
3.2 User-Producer Interactions
While evolutionary economics and business studies focus on the production-side and the creation of knowledge and innovation, with less attention to the user side, innovation studies focus their attention on the co-evolution of technologies and markets (Coombs et al., 2001). The adoption of medical technology is neither a passive nor a unidirectional. The diffusion of technological innovation responds to a series of interactions and feedback mechanisms between the users and the developers of a technology, with the demand and supply forces determining these feedback processes (Gelijns and Rosenberg, 1994).
Von Hippel proposed that innovation processes are distributed across users, manufacturers, suppliers and others, highlighting the importance of shifting from manufacturers-as-innovators into user-producer interactions as a source of innovation (Von Hippel, 1988). According to Von Hippel, these user-producer interactions control the survival of new technological artefacts in the market and ensure a demand for them (Von Hippel, 2005).
In medicine, these user-producer interactions often take place among clinicians who prescribe drugs to patients, patients who may report adverse events to clinicians, clinicians feeding this information to regulatory agencies and regulatory agencies or pharmacodiagnostics informing the manufacturers about safety and suspected adverse events.
3.3 Socio-technical systems
The diffusion of medical innovations is socially constructed and continuously negotiated between the members of the organisation (Greenhalgh, 2005). Medical innovations are embedded in a complex socio-economic environment formed by health organisations, institutions, regulations, communities of practitioners or patient organisations. This network explains how technology emerges, develops and translates into the clinic. This implies a co-evolution and a co-shaping of the technical as well as the socio-economic (Bijker and Law, 1992), therefore, any new medical technology will only reach the clinic when the complex elements that form its socio-technical network are such that enables the translation.
For the purpose of this study we considered that TPMT testing formed a socio-technical system composed of actors with different technological views and positions about the technology. These actors include researchers, clinicians, regulators, private companies, patient groups and policy-makers.
The core of this case study lies in understanding the evolution of the socio-technical network defined by TPMT testing.
4 Methods
The methods used in this case study were aimed at gathering arguments and opinions for and against the use of TPMT testing in order to inform its technological trajectory. The research methods were qualitative and were divided in two phases, first secondary research (document analysis) followed by observational research and interviews (primary research).
4.1 Document Analysis
A first exploratory document search gathered technical information on TPMT testing. The sources of evidence consulted were biological and medical journals, published socio-economic studies, reports, product brochures, newspaper articles and unpublished work. The desk research was also intended to look for secondary data that could support the information obtained, at a later stage during the interview process. Secondary data was obtained from government publications, regulations, clinical trial protocols and patents. All these documents provided empirical information about TPMT testing and illustrated ongoing debates around the use of the test.
4.2 Semi-Structured Interviews
The aim of the interview process was (1) validating the desk research and (2) obtaining views from key informants that provided reliable opinion about the benefits and hurdles of introducing TPMT testing in clinical practice. Interviewees were first selected from the literature and then followed a snowball sampling process where each interviewee appointed other experts.
Interviewees were clustered into groups of experts: researchers (formed by geneticists, biochemists, pharmacologists, medical researchers, health economists and sociologists), clinicians (haematologists, oncologists, gastroenterologists and rheumatologists) and regulators. Interviews were tailored to each of these groups of experts. A total of sixteen interviews was undertaken. Seven interviewees were life science researchers, all of them involved in TPMT testing, in either research only or research and service provision (in the case of NHS laboratories). Three were social science researchers with expertise in TPMT testing, four were clinicinas (one rheumatologist, two haematologists and a gastroenterologist) and another four were experts in personalised medicine not specific to TPMT testing. One commercial company involved in a test (not related to TPMT testing) was also interviewed as well as two respondents with a regulatory affiliation. These were involved in regulating pharmaceuticals and, although they had extensive knowledge about PM, they were not directly involved in the TPMT testing case.
A patient organisation did not agree for an interview as it considered PM was not relevant for them. Three other patient organisations did not respond.
5 TPMT Testing in the UK
TPMT testing is a pharmacodiagnostic tool that determines the levels of the enzyme Thiopurine Methyltransferase (TPMT) in the blood. This test predicts the likelihood of experiencing a serious or very serious adverse event to the thiopurine drugs azathioprine and 6-mercaptopurine, both off patent.
Thiopurines (Azathioprine-Imuran® and 6-Mercaptopurine-Purinethol®) are immunosuppressants used, since they were first marketed in the late 1950s by Wellcome (later on Glaxo and now GSK), for treating patients undergoing organ transplant surgery. Although these drugs were firstly aimed at avoiding transplant rejection, they were later on used to treat autoimmune conditions, mainly in dermatology (dermatomyositis, pemphigus vulgaris), rheumatology (systemic lupus erythematosus) and gastroenterology (Chron’s Disease, Ulcerative Cholitis), as well as in haematology to treat acute lymphoblastic leukaemia. Thiopurines, as well as any other drugs, have associated side-effects, principally a reduction in the production of blood cells that can seriously compromise the patient’s health. As a consequence, patients treated with these drugs need to be closely monitored (full blood count, liver function tests and electrolyte analysis) on a weekly basis, until the treatment is stabilised.
Some of the adverse events caused by thiopurines have been associated with low levels of or the lack of the enzyme Thiopurine Methyltransferase (TPMT) (Black et al., 1998, Arenas et al., 2006). According to the levels of the enzyme in the blood, patients can be advised not to take the drug or be prescribed a lower dose; however, the response to azathioprine has also a genetic component and, mutations in the gene that codes for the TPMT gene can also be associated with adverse events (Coulthard et al., 2004, Coulthard and Hogarth, 2005, Arenas et al., 2006). However, the correlation between enzyme levels (phenotype) and mutations (genotype) is not fully understood and testing for the enzyme levels is more effective in most cases than looking for genetic mutations. Only some of the mutations implicated in drug response are known.
For ALL the situation was different. TPMT testing was part of a clinical trial ALL, where every child diagnosed with ALL underwent TPMT testing. The major benefit of TPMT testing (phenotypic test) is diagnosing who is at risk of a severe ADR, which may be fatal. It is estimated that only 0.3% of the population might be exposed to that level of risk, 10% of the population might be at a moderate risk (not as severe) and the remaining 90% may develop a normal drug response. In the UK there are two NHS reference laboratories that offerTPMT testing although the test is not extended across the clinical community (Farguer et al., 2006). There are various reasons for this:
- TPMT testing in the UK is a laboratory developed or home-brew test. It is not a commercial kit as it does not comply with the In Vitro Diagnostics (IVD) Directive that rules the marketing of commercial diagnostic tests. On the contrary, it is a non-commercial test developed by NHS laboratories.
- The National Institute of Clinical Excellence (NICE), who is the main reimbursement body in the UK and decides which drugs and technologies should be adopted by the NHS, has not appraised the test. As a consequence, TPMT testing is not reimbursed at a central level. Individual primary care consortia (former Primary Care Trusts) may reimburse TPMT testing at a local level if they consider testing useful and necessary.
- In the lack of a formal evaluation of the test, there are different patterns of uptake between dermatologists, rheumatologists, gastroenterologists and haematologists (Payne et al., 2007). The British Society of Dermatologists considers that TPMT testing should be considered before prescribing azathioprine (Anstey et al., 2004), the British Society of Rheumatology has also adhered to this recommendation (Payne et al., 2007); however, the British Society of Gastroenterologists does not recommend TPMT testing as it considers that azathioprine has been widely used in Ulcerative Colitis and Crohn’s Disease and has proved to be safe (Teml et al., 2007).
6 TPMT testing in the UK Reimbursement System
The translation of any drug or medical technology into clinical practice requires a demonstration of comparative safety, efficacy and cost-effectiveness, together with other factors like disease severity as well as other ethical, social, and legal implications (Shah, 2004).
In the UK, NICE is the official body that decides on reimbursement at a national level. NICE evaluates primarily drugs but, since 2009 it also started evaluating diagnostics. NICE has a pragmatic view on how its decisions should be made and only recommends treatments below the threshold of £30,000 per Quality-Adjusted-Life Years (QALY).
TPMT testing emerged as a tool to predict adverse events originated by two off-patent drugs: AZA and 6-MP. Both AZA and 6-MP are reimbursed by the NHS because both drugs had been used before NICE started appraising technologies and because both are off-patent and therefore cheap drugs. The drugs cost approximately £20 a month (data provided by clinicians in 2007. The 2013 BNF cost of a pack of 28 pills of 25 mg is £6.02 .), while the test, £27 per patient (costs for 2007). But despite AZA is reimbursed by the NHS, TPMT testing is not, because NICE has not appraised the test and, without a “stamp” of approval that assures clinical utility, it is difficult for the NHS to justify reimbursement. Nevertheless and, despite TPMT was not appraised by NICE, the two National Reference Laboratories (at Birmingham City Hospital and Guy´s Hospital in London) offer TPMT testing to the NHS since 2003. As a result, some hospitals reimburse the test locally, depending on the cash balance of the NHE Trust or hospital as well as on other factors such as the clinician’s willingness to prescribe the test or their proximity or connections with the testing laboratory.
6.1 Service Delivery
Once a TPMT test is requested by a clinician, the patient signs a consent form and a sample is sent to one of the reference laboratories. The laboratory sends a report back within 6 working days, with a narrative interpretation, in which they assign a high or low risk of myelotoxicity and warn of the need for cautious use of AZA or 6-MP, although the report is only a recommendation. Depending of the levels of TPMT in blood, patients can be classified as:
- Low risk: the patient is normal (his/her TPMT levels are normal or even higher than normal)
- Higher risk than normal (TPMT levels are low) and azathioprine should be taken with caution (clinicians often reduce the dose by half).
- High risk (levels of TPMT are very low or nonexistent) and should not be given azathioprine.
Clinical demand varies across specialties. During the first year of service, referrals to Birmigham City Hospital came from the following clinical specialties: gastroenterology (66.7%), dermatology (13.6%), rheumatology (12%) and other specialties (7.7%) (Graham et al., 2004), with demand increasing in the following years. By diseases treated, TPMT testing was requested in patients with Crohn’s Disease (27.5%), Ulcerative Colitis (31.9%), Inflammatory Bowel Disease (4.8%), Systemic Lupus Erythematosus (4.4%), Dermatitis/Eczema (7.2%), Bullous Pemphigoid (6.3%) and others (7.6%) (Graham et al., 2004). It should be noted here that the high demand in gastroenterology is due to the high volume of patients in this specialty taking azathioprine, which outweighs the lack of recommendation for testing in gastroenterology.
7 Drivers for TPMT testing uptake
The main factors influencing testing uptake are the following:
- Promoters of TPMT testing argue that, the benefits of testing are substantial, not only for patients who have low or no TPMT levels and should not be taking the drug, but also for people with intermediate levels, who could benefit from a reduction in the AZA dose.
- The reference laboratories suggest that TPMT testing is cost-effective on the basis that it prevents serious AEs, which treatment costs are very high. Some studies have shown that TPMT enzyme test is also cost-effective in certain situations (Payne et al., 2009, Graham et al., 2004, Gurwitz et al., 2009).
- Some clinical groups have recommended TPMT testing. The British Association of Dermatology was the first group to recommended testing, as a result, all dermatologists across the UK now refer their patients for a test before prescribing AZA (Anstey et al., 2004). Subsequently, the British Society of Rheumatologists also included TPMT testing as an option to patients on AZA. As a result, the demand for test increased among rheumatologists (Payne et al., 2007).
- In 2007, some hospitals in the UK (approximately 20 NHS Trusts) have established TPMT prescreening policies, meaning that they have implemented TPMT testing and cover the cost of testing.
- Some respondents believe TPMT testing may prevent bone marrow failure in ALL patients, particularly when routine blood tests could not detect it.
- Phenotyping or measuring TPMT enzyme levels in blood appears to be the best form of testing. The enzyme assay gives more information than the genetic assay, which only looks at the most common polymorphisms that affect drug response. But not all polymorphisms that influence drug response are known and even if they were, doing a genetic analysis of all of them would be too costly and too lengthy. For this reason, genotyping is an option, only when phenotyping does not give a conclusive indication.
8 Barriers for TPMT testing uptake
Even though TPMT testing is already available at the NHS, there are a number of reasons why the diffusion of the test is slow and remains, in some instances controversial:
- The clinical opinions about testing are contradictory. While some haematologists consider the test effective and necessary, others believe that TPMT testing would not replace routine blood monitoring and electrolyte analysis.
- Not all clinical associations believe the test has an additional benefit. The British Society of Gastroenterology has never included TPMT testing in its recommendations (Carter et al., 2004).
- There is not a formal evaluation of TPMT test and, the lack of consensus on TPMT testing across specialties makes the situation complicated. It is not clear whether not doing a TPMT test could incur in malpractice.
- Neither of the TPMT cost and economic studies studies have been considered in any of the decisions on TPMT testing.
9 Discussion
TPMT testing exemplifies a case where PM can improve the efficacy of licensed drugs. Even thoughTPMT was one of the first biomarkers of drug response discovered and the indications that testing could predict serious AEs, the test has not been added to the product license of AZA or 6-MP.
In 2003, the Department of Health (DoH) published a white paper announcing funding for research in PM and genetics and encouraging researchers to look for specialised service delivery mechanisms. According to this white paper, “…patients could undergo a test to predict their response or ensure medicine and doses are right at the first time”. This was particularly relevant to patients at risk of AEs as, only in the UK, AEs were estimated to affect 7% of hospital admissions at an annual cost of £380 million to the NHS in England alone (DoH 2003).
In addition, the DoH added that “…the logistics and clinical utility of including a test in prescribing decisions will need careful evaluation, but applying this knowledge within primary care should significantly improve patient outcomes in medicines use” (DoH 2003). However, to date, TPMT testing has not received a careful evaluation by NICE or any other UK reimbursement body, even if TPMT is a well known biomarker and testing facilities are available at the NHS.
Since NICE launched its diagnostics assessment programme in 2009, evaluations have focused on commercial diagnostics rather than home-brew tests (such as TPMT testing), even if these addressed an unmet need. This verifies the hypothesis that, the UK evaluation system for medical devices is not designed to address products for an unmet need, if these do not have business potential. But, despite the lack of institutional support, TPMT testing has been introduced in clinical practice through alternative routes. As we have seen in the drivers and barriers sections, the demand for testing increased considerably after two reference laboratories started offering testing (Ford et al., 2004a, Ford et al., 2004b, Birmingham City Hospital, 2009), followed by clinical associations including TPMT testing in their guidelines.
TPMT testing reference laboratories acted as lead users of the test, setting a national service across the whole NHS. These laboratories were also successful at optimising the technology for measuring the levels of enzyme in the blood and disseminating information about the benefits of testing.
The fact that two public laboratories have taken the lead in the UK and are offering TPMT testing services, indicates that:
- Expertise in product and service innovation in PM and diagnostic testing is not exclusive to the diagnostic or pharmaceutical industry. Other actors like public laboratories are important innovators, particularly in non-commercial diagnostics.
- The case of TPMT testing shows that, the implementation of PM for preventing AEs is strongly driven by lead users who reside in public hospitals. Clinicians and public laboratories are key actors designing the technological trajectories of PM for improving drug safety.
- PM for off-patent drugs is strongly associated to home-brew tests developed in NHS laboratories. However, private companies could threaten public service provision, if other commercial alternatives emerged and/or if alternative commercial tests were reimbursed.
- Peer opinion exerts a strong influence on clinical acceptance and demand for testing. Professional guidelines are strong drivers for technology adoption, for this reason, even though TPMT testing had not been appraised by NICE, it has been recommended by some clinical associations and requested by certain clinics and hospitals.
10 Conclusion
Market access for drugs and commercial diagnostics lies on the demonstration of safety, efficacy and cost-effectiveness, assessed by regulatory and reimbursement agencies. The case of TPMT testing, however, shows that PM for off-patent drugs is associated to non-commercial diagnostics. These do not follow the same evaluation procedures than commercial tests. The case of TPMT testing shows that, process of diffusion of these home-brew tests lies in public laboratories that generate testing capabilities and disseminate knowledge across the clinical community.
Acknowledgements:
I thank Professor Denis Loveridge and Dr. Michael Keenan for their support in this work as well as the Manchester Institute of Innovation Research for financial support.
References
Anstey, A. V., Wakelin, S., Reynolds, N. J. (2004): Guidelines for prescribing azathioprine in dermatology, British Journal of Dermatology, 151, 1123-1132.
Arenas, M., Marinkaki, A., Ansari, A., Sanderson, J. (2006): Typing TPMT and ITPase to detect azathioprine toxicity, Personalised Medicine, 3, p. 45-59.
Arenas, M., Simpson, G., Lewis, C. M., Shobowale-Bakre, E.-M., Escuredo, E., Fairbanks, L. D., Duley, J. A., Ansari, A., Sanderson, J. D., Marinaki, A. M. (2005): Genetic Variation in the MTHFR Gene Influences Thiopurine Methyltransferase Activity, Clin Chem, 51, p. 2371-2374.
Arnold, H. P., Hall, S. T. (2005): Pharmacogenomics and Clinical R&D, Pharmacogenomics, 6, p. 801-806.
Bijker, W. E. (1995): Of Bicycles, Bakelites and Bulbs, Cambridge – Massachusetts The MIT Press.
Birmingham City Hospital (2009): Thiopurine S-methyl transferase (TPMT) Service.
Black, A. J., McLeod, H. L., Capell, H. A., Powrie, R. H., Matowe, L. K., Pritchard, S. C., Collie-Duguid, E. S. & Reid, D. M. (1998): Thiopurine methyltransferase genotype predicts therapy-limiting severe toxicity from azathioprine, Annual International Medicine, 129, p. 716 718.
Carter, M. J., Lobo, A. J., Travis, S. P. L. (2004): Guidelines for the management of inflammatory bowel disease in adults, On behalf of the IBD Section of the British Society of Gastroenterology, International Journal of Gastroenterology and Hepatology, 53, v1-v6.
Coleman, J. S., Katz, E., Menzel, H. (1966): Medical innovation : a diffusion study, Indianapolis, Bobbs-Merrill.
Coombs, R., Green, K., Richards, A., Walsh, V. (Eds.) (2001): Technology and the Market: Demand, and Innovation, Cheltenham, UK, Edward Elgar.
Coulthard, S., Hogarth, L. (2005): The thiopurines: An Update, Investigational New Drugs, 23, p. 523-532.
Coulthard, S. A., Matheson, E. C., Hall, A. G., Hogarth, L. A. (2004): The Clinical Impact of Thiopurine Methyltransferase Polymorphisms on Thiopurine Treatment, Nucleoside, Nucleotides and Nucleic Acids, 23, p. 1385-1391.
Di Masi, J., Hansen, R. W., Grabowski, H. G. (2003): The price of innovation: new estimates of drug development costs, Journal of health Economics, 22, p. 151-185.
DOH (2003): Government White Paper “Our inheritance, our future: realising the potential of genetics in the NHS”, Department of Health.
Dosi, G. (1982): Technological Paradigms and technological trajectories, Research Policy, 11, p. 147-162.
Evans, W., Johnson, J. (2001): Pharmacogenomics: the inherited basis for interindividual differences in drug response, Annual Review of Genomics and Human Genetics, 2, p. 9-39.
Fargher, E., Tricker, K., Newman, B., Elliott, R. A., Roberts, S., Shaffer, J., Bruce, I., K., P. (2007): Current use of pharmacogenetic testing: a national survey of thiopurine methyltransferase testing prior to azathioprine prescription, Journal of Clinical Pharmacy and Therapeutics, 32, p. 187-95.
Farguer, E., Tricker, K. J., Newman, W., Elliot, R. A., Ollier, W. E. R., Shaffer, J., Griffiths, C. E., Bruce, I., Payne, K. (2006): Pharmacogenetic testing for azathioprine in the NHS: current uptake and implications for prescribing practice, The International Journal of Pharmacy, Supplement 2, B70-1.
FDA (2004): Innovation and Stagnation, Challenge and Opportunity on the Critical Path to New Medical Products
Ford, L., Graham, V., Berg, J. (2004a): Individualising therapy; a new whole blood phenotypic assay for TPMT and concordance with genotyping, Sandwell and West Birmingham Hospitals NHS Trust.
Ford, L., Graham, V., Berg, J. (2004b): Patients with high TPMT activity identified during routine phenotypic testing-what is going on?, Sandwell and West Birmingham Hospitals NHS Trust.
Gelijns, A. & Rosenberg, N. (1994): The dynamics of technological change in medicine, Health Affairs, 13, p. 28-46.
Graham, V., Ford, L., Berg, J. (2004): Twelve Months of Referral Service. Has it made a difference?, Sandwell and West Birmingham Hospitals NHS Trust.
Greenhalgh, T., Robert, G., Macfarlane, F., Bate, P., Kyriakidou, O. (2004): Diffusion of Innovations in Service Organizations: Systematic Review and Recommendations, The Milbank Quarterly, 82, p. 581-629.
Gurwitz, D., Rodriguez-Antona, C., Payne, K., Newman, W., Gisbert, J. P., Mesa, E. G. D., Ibarreta, D. (2009): Improving pharmacovigilance in Europe: TPMT genotyping and phenotyping in the UK and Spain, European Journal of Human Genetics, 17.
Hedgecoe, A., Martin, P. (2003): The Drugs Don’t Work: Expectations and the Shaping of Pharmacogenetics, Social Studies of Science, 33, p. 327-364.
Human Genetics Commission ( 2002): Information-Gathering Session on Pharmacogenetics
Lewis, G. (2003): The Clinical and Commercial Development of Pharmacogenetics, Project Outline.
Lindpaintner, K. (2003): The impact of pharmacogenetics and pharmacogenomics, Journal of Commercial Biotechnology, 10, p. 60-77.Milne, C.-P. (2002): Orphan products—pain relief for clinical development headaches, Nature Biotechnology, 20, p. 780-784.
Medical Research Council (2003): UK ALL 2003 Protocol, UK National Randomised Trial for Children and Young Adults with Acute Lymphoblastic Leukaemia (ALL).
Need, A., Motulsky, A. G., Goldstein, D. B. (2005): Priorities and standards in pharmacogenetic research, Nature Genetics, 37, p. 671-681.
NICE (2009): Measuring effectiveness and cost effectiveness: the QALY, The National Institute of Clinical Excellence.
Payne, K., Newman, W., Farguer, E., Tricker, K., Bruce, I. N., Ollier, W. E. R. (2007): TPMT Testing in rheumatology: any better than routine monitoring?, Rheumatology.
Payne, K., Newman, W. G., Gurwitz, D., Ibarreta, D., Phillips, K. A. (2009): TPMT Testing in azathioprine: a “cost-effective use of healthcare resources”?, Personalized Medicine, 6, p. 103-13.
Rogers, E. M. (1962): Diffusion of Innovations, New York, The Free Press.
Shah, J. (2004): Criteria influencing the clinical uptake of pharmacogenomic strategies, British Medical Journal, 328.
Shah, R. R. (2006): Can pharmacogenetics help rescue drugs withdrawn from the market?, Pharmacogenomics, 7, p. 889-908.
Teml, A., Schaeffeler, E., Herrlinger, K., Klotz, U., Schwab, M. (2007): Thiopurine Treatment in Inflammatory Bowel Disease: Clinical Pharmacology and Implication of Pharmacogenetically Guided Dosing Clinical Pharmacokinetics, 46, 187-208.
Von Hippel, E. (1988): The Sources of Innovation, New York, Oxford, Oxford University Press.
Von Hippel, E. (2005): Democratizing Innovation, The MIT Press.
Webster, A., Martin, P., Lewis, G., Smart, A. (2004): Integrating pharmacogenetics into society: in search of a model, Nature Reviews Genetics, 5, p. 663-668.